The ear length is measured using electronic calipers from the superior helix to the inferior tip of the lobe and can be measured in the coronal or sagittal plane on 2D imaging and on sagittal plane on 3D imaging.
Abnormalities of the auricle’s size and number
Author: Ian Suchet1, Janelle Santos2
1. Ian Suchet MBBCh FRCPC, Calgary MFM Centre, EFW Radiology, Calgary, Alberta Canada
2. Mayo Clinic, Department of Obstetrics and Gynecology, Rochester, MN, USA
Reviewers: Karen Fung-Kee-Fung, Mauro Schenone
Normal size of the auricle
Ear length (Table 2 and 3), width and area have been shown to correlate with increasing gestational age (1-4).
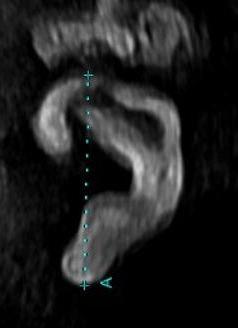
The ear length is measured using electronic calipers from the superior helix to the inferior tip of the lobe and can be measured in the coronal or sagittal plane on 2D imaging and on sagittal plane on 3D imaging (Figure 15).
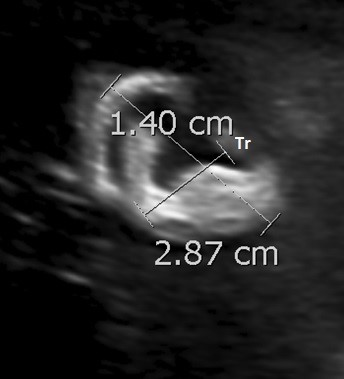
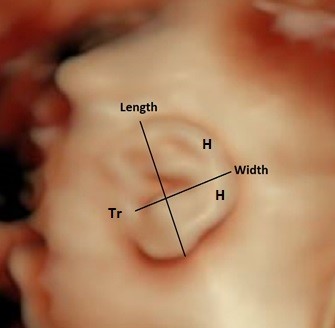
The ear width is measured by placing the caliper at the most posterior edge of the tragus and plotting a right-angled line to the ear length measurement, ending at the outermost aspect of the helix (Figure 15).
I have found that 2D imaging underestimates the size of the auricle by as much as 10% (unpublished data) as it is difficult to accurately delineated the exact superior extent of the helix and inferior extent of the lobe on the single 2D image (Figure 16).
Numerous normal nomograms have been created (1).
Other ratios that have been reported include:
- Ear length is 1/3 BPD.
- Length / width ratios have been established. The median is 1.9 with a slight decline over the gestational period (2). These workers believe that this ratio could prospectively be used as a marker in syndromic detection (5). Ear length measurements have been used to assess for fetal aneuploidy (4-6).
|
Table 2: Nomograms for external ear length. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Joshi KS, Chawla CD, Karki S et.al. Sonographic measurement if fetal pinna length in normal pregnancies. Kathmandu University Medical Journal 2011;9:49-53.
Figure 15. Ear length and width measurements in a sagittal plane.
a,b – 2D ultrasound.
c,d – 3 D ultrasound.
Tr – tagus; H – helix.
A
B
C
D
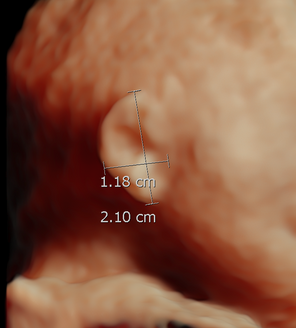
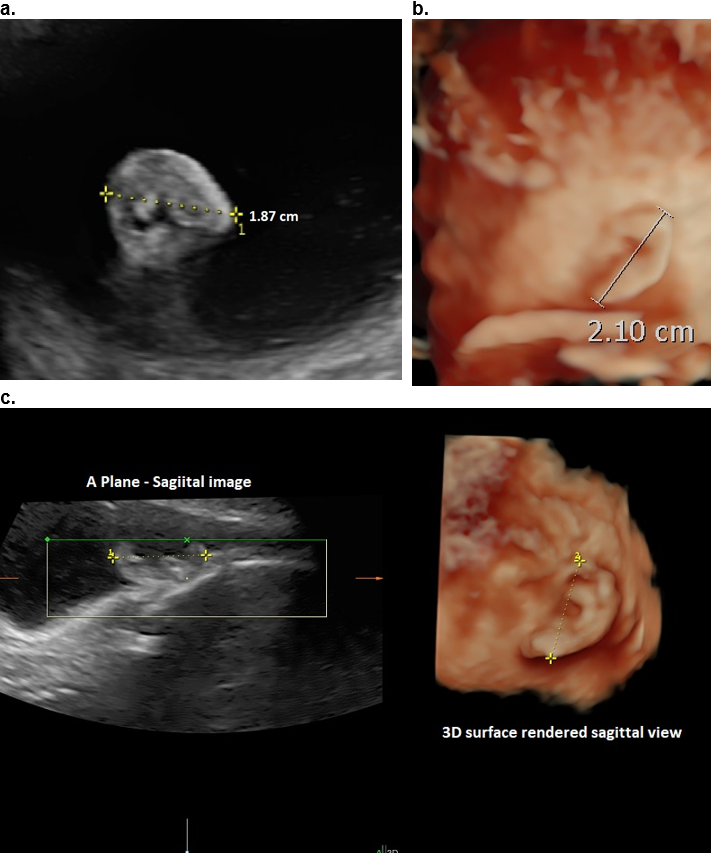
Figure 16. 24 wks GA. 2D ultrasound versus 3D ultrasound for ear length measurement on the same patient
a. 2D sagittal image – length 1.87 cm,
b. 3D image on the same fetus 2.1 cm.
c, 3D MPR on another fetus. Similar but smaller measurement on the sagittal A plane image (left panel) when compared to the 3D surface rendered image (right panel).
References
- Chitkara U, Lee L, Li-Sayed YV, er al. Ultrasonographic ear length measurement in normal second and third trimester fetuses. Am J Obstet Gynecol 2000;183:230-234.
- Sondern K, Kretiz K, Hammer K et.al. 3D ultrasound evaluation of the fetal outer ear: Novel biometric ratio and comparison of different surface display modes. Fetal Diagn Ther 2019;46:200-206.
- Dudarewicz L, Kałuzewski B. Prenatal screening for fetal chromosomal abnormalities using ear length and shape as an ultrasound marker. Med Sci Monit. 2000; 6(4): 801–806.
- Chang CH, Chang FM, Yu CH, Liang RI, Ko HC, Chen HY. Fetal ear assessment and prenatal detection of aneuploidy by the quantitative three-dimensional ultrasonography. Ultrasound Med Biol. 2000; 26(5): 743–749.
- Hatanaka AR, Rolo LC, Mattar R, Araujo Junior E, Nardozza LM, Moron AF. Reference intervals for fetal ear length between 19 and 24 weeks of pregnancy on 3-dimensional sonography. J Ultrasound Med. 2011;30(9): 1185–1190.
- Chitkara U, Lee L, Oehlert JW, Bloch DA, Holbrook RH Jr, El-Sayed YY, et al. Fetal ear length measurement: a useful predictor of aneuploidy? Ultrasound Obstet Gynecol. 2002; 19(2): 131–135.
Microtia / Anotia
Definition
Microtia is a broad term that encompasses a diverse array of malformations of underdevelopment of the auricular components of the ear. The malformations range from a small but normal auricle to mild structural abnormalities of the auricle to hypoplasia or complete absence of the auricle and EAC (anotia) at the other end of the spectrum (1-6).
Congenital microtia is one of the most common surface birth defects. It is the second most common facial deformity after cleft lip and palate.
Incidence
0.8–17.4 per 10,000 live births (7,8). Hispanics and Asians have a higher prevalence than African Americans and Europeans. A high incidence of 12 per 10,000 live births has been reported in Navajo Indians (9). The prevalence in Finland is 4.34/10,000 (10). The highest rates are present in South America with rates of 1:500-3000. Quito has the highest rate of 1:500 (11).
Classification Systems for Microtia
The phenotypic variability of congenital anomalies of the ear makes the development of a universally accepted classification system challenging. Numerous classification systems have been developed to facilitate both the diagnosis and treatment of microtia, as well as to allow for standardized data collection in multi-center studies.
Hermann Marx (12) published the first system, the Marx classification, in 1926 and it remains one of the most frequently used systems.
Tanzer (13) classification of ear abnormalities correlates with the surgical approach.
Weerda (14) modified the Marx and Tanzer definitions based on embryologic development as well as surgical steps and included all congenital abnormalities of the external ear (i.e., deformities and minor anomalies).
The American Journal of Medical Genetics has recently published a collection of articles in an effort to standardize external ear terminology in the clinical genetics field (15). The Weerda classification system was chosen as the basis for the standardized terminology used for microtia. These classification systems, commonly cited in studies of microtia, are summarized in Table 4.
Ultrasound
Microtia is diagnosed by at least both decreased longitudinal length (2 SD below the mean) and width, and in more severe forms it includes an abnormal shape of auricle structure. Sonographically it presents as an abnormal size of the auricle, with / without an abnormal shape and / or position of the auricle in more severe cases (see Table 5).
- Unilateral in 77–93% of cases (10,16). In unilateral microtia, the location is right sided in 60% of cases.
- In larger series, about 10% of individuals were affected bilaterally, and about 80% had meatal atresia (16).
- Male > female (16). Epidemiologic studies have consistently shown a male to female predominance of about 2:1.
- Microtia can occur as an isolated anatomical anomaly or as part of a syndrome. Of the cases that appear isolated on an antenatal scan, postnatally, 20-40% of affected children (depending on the study) have been shown to have either a recognizable syndrome or at least one associated anomaly not directly related to the ear abnormality (17).
- Bilateral microtia is more likely to have associated anomalies (18) especially Treacher Collins syndrome and Goldenhar syndrome (18).
- Although microtia specifically refers to abnormalities of the auricle, over 90% of cases have stenosis or atresia of the EAC, leading to conductive hearing impairment requiring complex postnatal surgery. There may also be associated abnormalities of the tympanic membrane, middle ear ossicles or a combination of these (19).
- Prenatal detection of patency of the EAC by ultrasound is very low as only the fluid filled external opening. the EAM, may be visualized. The complete canal is obscured by normal ossification of the temporal bone. As the EAC is fluid filled, this acts as a natural contrast agent when using MRI resulting in far better visualization (20-22).
Frequency of microtia types: In a surgical series of 311 cases, Type 1 - 18%, type 2 - 20%, type 3 - 50%, type 4 - 3%. The reminder of the cases had meatal atresia without external ear defects (5). In larger population series, anotia represents 10-20% of cases (16).
Figure 17. 28 wks GA.Type 1 Microtia in two different patients. (GA 35 wks and 18 wks respectively). Small, low set ears. Auricular anatomy normal. EAC was patent.
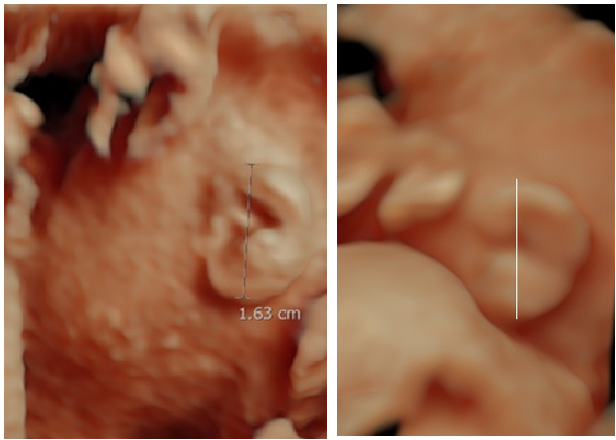
Figure 18: Type 2 Microtia.
a. Case 1 (20 wks GA): Small normally located ear. The auricle is partially intact but still has the appearance of an auricle. Stenosis of the EAC.
b. Case 2 (19 wks GA): Small low set ear. Auricle intact. EAM was found to be absent / atretic on postnatal evaluation.
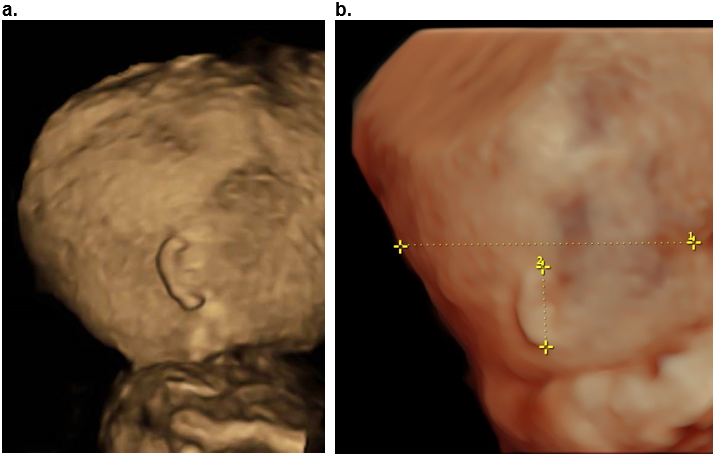
Figure 19. Type 3 Microtia.
a, 3D surface rendered view (a) and postnatal image (b) of the left ear at 22 wks of gestation. Vertical mass of tissue that does not resemble a normal ear. No normal auricular anatomy present. EAC atresia.
b. Postnatal image in the same patient.
c,d. MRI at 29 wks GA in another patient.
c. Coronal plane. Normal right ear. Small left ear on the coronal image.
d. The left IAC has some fluid within it (closed arrows) however there is an area of stenosis noted in midportion (open black arrow).
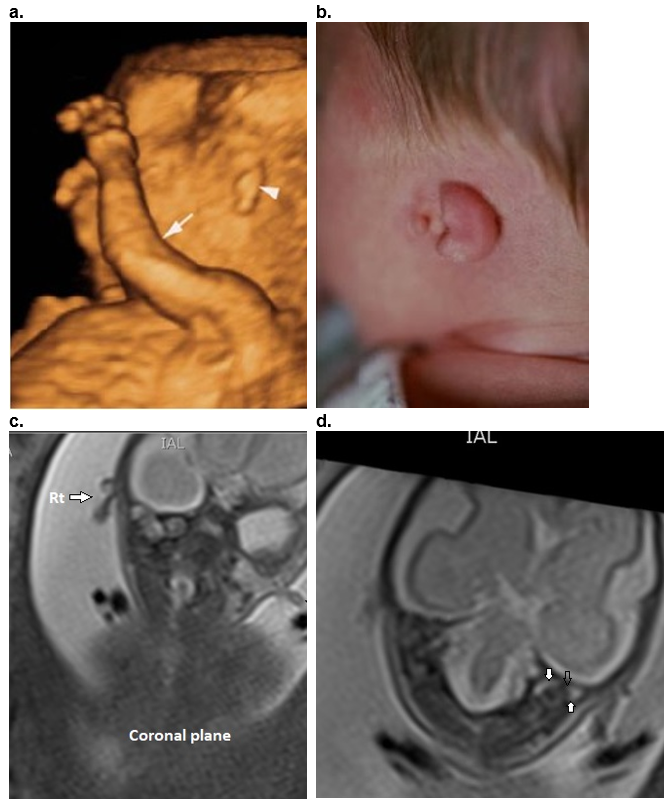
Figure 20. Type 4 Bilateral Microtia – 3D surface rendered views. Rudimentary tissue present bilaterally with no visible normal auricular tissue. Complete atresia of EAC bilaterally.
a. Left ear.
b. Right ear in the same patient.
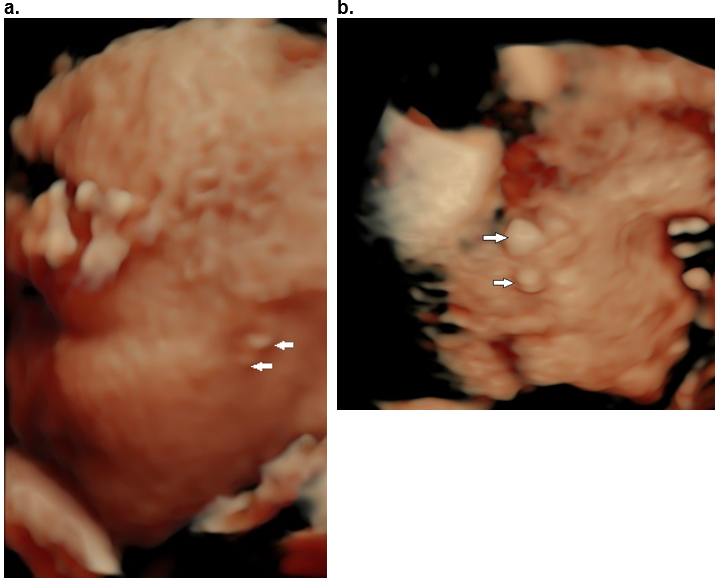
Figure 21. Trisomy 13. Bilateral anotia. There is also bilateral anophthalmia and a small mouth (microstomia).
A. 3D surface rendered view at 20 weeks of gestation demonstrating an absent ear (arrow).
B. Postabortal images of the same patient.
Risk factors and associated syndromes
In two published large population series, 28-49% of reported cases had microtia associated with other defects, with only 6-9% having a well- defined syndrome (3-5).
Most cases of microtia/anotia are isolated, with no family history, and most large surgical series indicate that less than 10% of the time there is a family history.
In two published large population series, 28-49% of reported cases had microtia associated with other defects, with only 6-9% having a well- defined syndrome (3-5).
Numerous authors have mentioned the occasional occurrence of a positive family history in non syndromic microtia.
Rollick and Kaye (23) have documented higher frequencies of first- and second-degree relatives with ear abnormalities in individuals with microtia with or without hemifacial microsomia. In their studies, 8% of first-degree relatives had some degree of ear abnormality, including preauricular tags. While most of their patients were classified as having hemifacial microsomia as opposed to isolated microtia, eight of 15 of their cases diagnosed as microtia had a positive family history. Their data suggest that microtia is part of a continuum that extends to hemifacial microsomia and Goldenhar syndrome.
The most important condition associated with microtia is the oculo-auriculo-vertebral spectrum (OAVS).
Investigators at the Center for Craniofacial Anomalies at the University of Illinois (23) documented the relationship between isolated microtia, ear tags, hemifacial microsomia, Goldenhar syndrome, and the OAV spectrum. The range of findings within familial cases of the Goldenhar syndrome and of isolated (apparently non syndromic) microtia suggests a continuum; however, it is not clear that all individuals who have isolated microtia always have it as one end of the OAVS.
Llano-Riva et al. (25) studied 145 children with microtia, many having features of OAVS. Because of no difference in the groups of patients, they concluded that microtia is the mild end of expression of the OAVS. They acknowledge that not all of the syndromes in which microtia occurs will exhibit the entire spectrum.
Gorlin et al. (26) and Cohen et al. (27) have presented reviews of the OAVS. It is clear from all his work that this malformation pattern is a condition of etiologic and pathogenetic heterogeneity. Any individual who has apparently isolated microtia (with or without hemifacial microsomia) is a candidate for this diagnosis.
Microtia and OAVS share the following characteristics:
1) variable phenotypic expression,
2) asymmetric involvement of facial structures,
3) right side preponderance,
4) male predilection, and
5) familial occurrence of microtia or related anomalies such as preauricular tags and pits.
Based on these observations, it has been suggested that isolated microtia represents a milder phenotype of OAVS (7, 23, 25). This has led to the controversial concept that most (or all) cases presenting with apparent isolated microtia are actually cases of OAVS.
Other reported risk factors for microtia include:
- maternal diabetes mellitus,
- low birth weight,
- advanced maternal age,
- exposure to thalidomide, retinoic acid derivatives, and mycophenolate mofetil (28,29).
- autosomal trisomies (13,18, 21, and 22) and chromosome 18 mosaicism.
- Single‐gene disorders, for example SIX1 and EYA1 and chromosomal translocation 6p24, are also reported to be associated with microtia.
- Others include, High parity (7), First parity, Multiple births, Race (7), Paternal age (8), male sex, Low birthrate, maternal acute illness, advanced maternal age, maternal exposure to altitude, and many others (2).
- The London Dysmorphology Database of the London Medical Databases (version 1.0.1, http://www.lmdatabases.com) contains information on nearly 4000 dysmorphic and multiple congenital anomaly syndromes. Using microtia as a key word there are about 150 syndromes or conditions in this database. Many of these conditions are very rare: Treacher Collins, Nager, and Branchio-Oto-Renal and Goldenhar syndrome are the most common.
About 20-40% of children with microtia/anotia will have an associated defect or an identifiable syndrome pattern (3,8,24).
Gorlin et al. (8) and Tewfik and der Kaloustian (30) have cataloged the many syndromes that may have microtia as a feature.
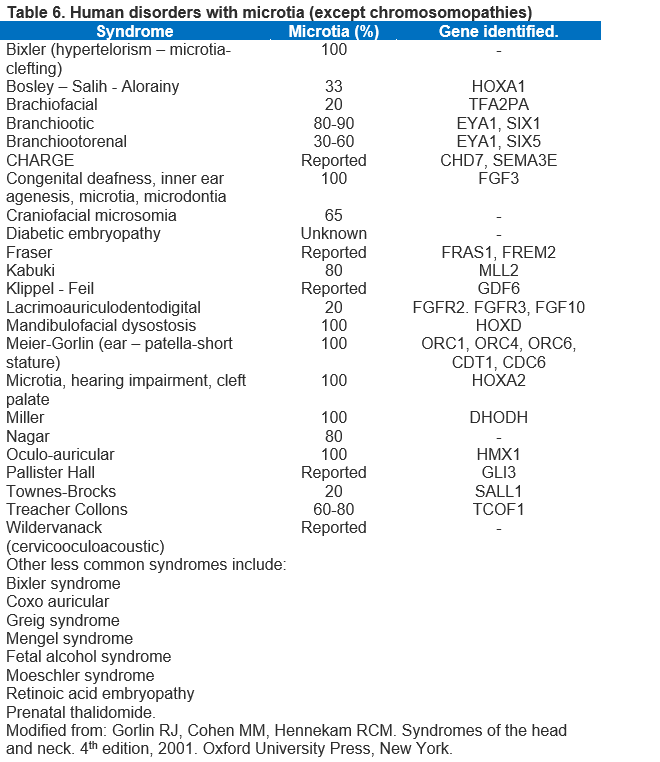
Table 6 lists some of the more common syndromes and genetics (Ref 1).
Other non-random associations
In addition to the list of well-delineated syndromes, microtia appears to have other, non-random associations.
- Kaye et al. (31) documented a positive association between microtia and cervical spine fusion not necessarily as part of the OAV spectrum. Their data suggest that individuals with isolated microtia should be investigated for the presence of cervical spine abnormalities.
- External ear defects and renal malformations. Other than the occurrence of auricular abnormalities (including microtia) in the many syndromes characterized by both ear and kidney defects, there is no conclusive epidemiologic evidence that an external ear defect is a marker for renal malformation and warrants invasive urologic investigation.
- The most important association with microtia is conductive and sensorineural hearing loss.
- Jaffe (32) suggested that the entire range of microtia as well as other milder ear defects should always suggest the possibility of associated middle and internal ear abnormalities.
- Bassila and Goldberg (33) found a 16% occurrence of sensorineural hearing loss in individuals with microtia and/or hemifacial microsomia.
- Jafek et al. (34) found no cases had neural hearing loss.
- Suutarla et al. (10) Found aural atresia or stenosis in 93% of patients, conductive hearing loss in 96% and sensorineural hearing loss in 8%of affected ears in Finland.
Inner ear abnormalities and sensorineural hearing loss should be excluded when following individuals with microtia.
References
- Deep A, Mortazavi MM, Adeep N. Ear. Bergman’s Comprehensive Encyclopedia of Human Anatomic Variation, First Edition. Edited by R. Shane Tubbs, Mohammadali M. Shoja and Marios Loukas. 2016 John Wiley & Sons, Inc. Published 2016 by John Wiley & Sons, Inc.
- Luquetti DV, Heike CL, Hing AV, Cunningham ML, Cox TC. Microtia: epidemiology and genetics. Am J Med Genet 2012;158:124–139.
- Kaye C, Rollick BR, Hauck WW, et al. Microtia and associated anomalies: statistical analysis. Am I Med Genet 1989;34:574.
- Bennun RD, Mulliken IB, Kaban LB, et al. Microtia: a microform of hemifacial microsomia. Plast Reconstr Surg 1985;76;859.
- Jafek BW, Nager GT, Strife I, et al. Congenital aural atresia: an analysis of 311 cases. Trans Am Acad Ophthalmol Otol 1975;80:588.
- Jahrsdoerfer R: Congenital malformations of the ear: analysis of 94 operations. Ann Otol Rhinol Laryngol 1980; 89:348.
- Tasse C, Bohringer S, Fischer S et al. Oculo-auriculo-vertebral spectrum (OAVS): clinical evaluation and severity scoring of 53 patients and proposal for a new classification. Eur. J. Med. Genet 2005; 48:397-411.
- Gorlin RJ, Jue KL, Jacobsen ULL et.al. Oculoauriculovertebral dysplasia. J. Pediatr. 1963;63:991-999.
- Nelson SM, Berry RI. Ear disease and hearing loss among Navajo children–a mass survey. Laryngoscope 1984;94:316–323.
- Suutarla S, Raution J, Ritvanen A et.al. Microtia in Finland: comparison of characteristics in different populations. Int J pediatr Otorhinolaryngol 2007;71:1211-1217.
- Castilla EE, Orioli IM: Prevalence rates of microtia in South America. Int J Epidemiol 1986;15:364.
- Marx H. 1926. Die Missbildungen des ohres. In: Denker A, Kahler O, editor. Handbuch der Spez Path Anatomie Histologie. Berlin, Germany: Springer. pp. 131
- Tanzer RC. Microtia. Clin Plast Surg 1978;5:317–336.
- Weerda H. Classification of congenital deformities of the auricle. Facial Plast Surg 1988;5:385–388
- Hunter A, Frias JL, Gillessen-Kaesbach G, Hughes H, Jones KL, Wilson L. Elements of morphology: Standard terminology for the ear. Am J Med Genet Part A 2009;149A:40–60.
- Stevenson RE, Hall JG. Human malformations and related anomalies. 2005. 2nd edition. Oxford University Press.
- Carey JC, Park AH, Muntz HR. External Ear. In: Stevenson RE, editor. Human malformations and related anomalies. Oxford University Press; Oxford; New York: 2006. pp. 329–338
- Heike CL, Hing AV. Craniofacial Microsomia Overview. In: Pagon RABT, Dolan CR, Stephens K, editors. Gene Reviews. 2010/03/20 ed University of Washington; Seattle: 2009.
- Takano K. Hearing loss in congenital microtia. Open access peer reviewed chapter 2017. DOI:10.5772/intechopen. 72429.
- Kaga K, Asato H. Microtia and atresia – combined approach by plastic and otologic surgery. Advances in Oto-Rhino-Laryngology 2014;Volume 75. Karger Publishers.
- Chang CC, Steinbacher DM. Treacher collins syndrome. Semin Plast Surg. 2012.
- Ishimoto S, ito K, Karino S et.al. Hearing levels in patients with microtia: correlation with temporal lobe malformation. Larygoscopy 2007;117:461-465.
- Rollick BR, Kaye C: Hemifacial microsomia and variants: pedigree data. Am J Med Genet 1983;15:233.
- Mastroiacova P, Corchia C, Bott LD, et al.: Epidemiology and genetics of microtia-anotia: a registry-based study on over one million births. ] Med Genet 1995;32:453.
- Llano-Riva I, Gonzalez-del Angel A, del Castillo V, et al.: Microtia: a clinical and genetic study at the National Institute of Pediatrics in Mexico City. Arch Med Res 1999;30:120.
- Gorlin RI, Cohen MM Ir, Hennekam RCM: Syndromes of the Head and Neck, ed 4. Oxford University Press, New York, 2001.
- Cohen MM, Rollick BR, Kaye CI, et al.: Oculoauriculovertebral spectrum: an updated critique. Cleft Palate 1989;126:276
- Anderka MT, Lin AE, Abuelo DN, Mitchell AA, Rasmussen SA. Reviewing the evidence for mycophenolate mofetil as a new teratogen: Case report and review of the literature. Am J Med Genet Part A 2009;149A:1241–1248.
- Klieger-Grossmann C, Chitayat D, Lavign S, et.al. Prenatal exposure to mycophenolate mofetil: An updated estimate. J Obstet Gynaecol Can 2010;32:794–797.
- Tewfik TD, der Kaloustian VM: Congenital Anomalies of the Ear, Nose,and Throat. Oxford University Press, New York, 1997.
- Kaye C, Rollick BR, Hauck WW, et al.: Microtia and associated anomalies: statistical analysis. Am J Med Genet 1989;34:574
- Jaffe BF. Pinna anomalies associated with congenital conductive hearing loss. Pediatrics 1976; 57:332.
- Bassila MK, Goldberg R: The association of facial palsy and/or sensorineural hearing loss in patients with hemifacial microsomia. Cleft Palate 1989;126:287
- S. Jafek BW, Nager GT, Strife I, et al. Congenital aural atresia: an analysis of 311 cases. Trans Am Acad Ophthalmol Otol 1975; 80:588.
Macrotia
Macrotia is an abnormal enlargement of the auricle: an ear with length and width for age both greater than 2 SD above the mean (Figure 22).
The normal ear is divided into three parts (see Anatomy section). In true macrotia, the upper third of the ear is most commonly enlarged. If the ear length is only increased, it should be called Long ear and not macrotia (1,2).
Etiology
(Adapted from: Stevenson RE, Hall JG. Everman B, Soloman BD. Human malformations and related anomalies. 3rd edition. Oxford University Press. 2015)
- Isolated (most cases).
- Marfans syndrome.
- Cerebro-oculo-facio-skeletal syndrome.
- Fragile X syndrome (long face, large testes, lax joints).
- Borjeson-Forssman-Lehmann syndrome (course face, hypogonadism).
- Langer-Gideon syndrome (microcephaly, exostoses, abnormal facial features).
- Melnick-Needles (distinctive face + skeletal changes).
- Pallister-Killian syndrome.
- Weaver syndrome.
- Nance-Horan syndrome.
- Chromosomal anomalies.
- Some workers believe that macrotia is an excessive overgrowth phenomenon. Oligohydramnios has been suggested as a pathogenetic mechanism, with intrauterine constraint inducing excessive ear growth (3).
Fragile X Syndrome (Figure 22)
- The auricles are very large (7 cm) in 70% of cases).
- The auricle is cupped with simple helices and absent lobes (4,5)
- The auricle may be everted.
- 25%-40% of carrier females, especially the more severely affected patients have the typical facies (long face, prognathism, large, everted pinnae) (6,7).
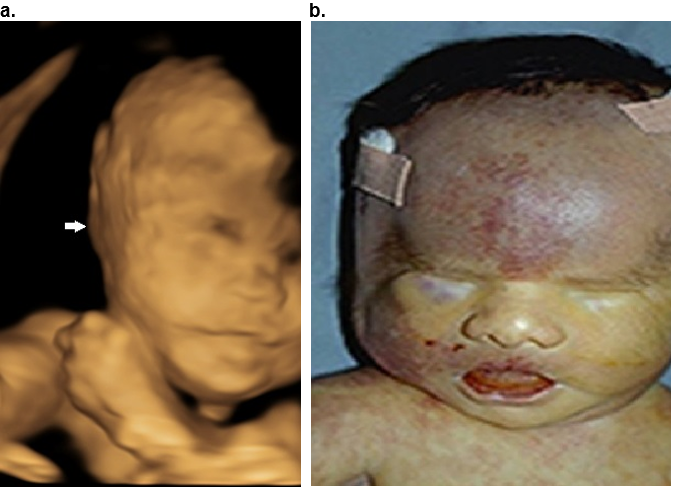
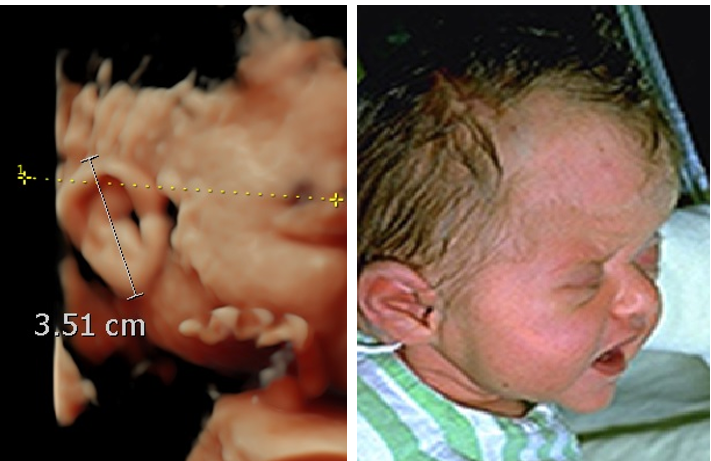
Figure 22. Macrotia. Fragile X syndrome at 26 weeks of gestation. The ear length is 35.1 mm (above the 95th percentile).
A. Sagittal surface rendered 3D image at 28 wks. Macrotia and slight posterior rotation of the auricle..
B. Postnatal image of the same patient.
References
- Deep A, Mortazavi MM, Adeep N. Ear. Bergman’s Comprehensive Encyclopedia of Human Anatomic Variation, First Edition. Edited by R. Shane Tubbs, Mohammadali M. Shoja and Marios Loukas. 2016 John Wiley & Sons, Inc. Published 2016 by John Wiley & Sons, Inc.
- Otto HD. Pathogenese der Aurikularanhänge, Melotie und Polyotie. Arch Otorhinolaryngol 1979;225(1):45-56.
- Aase JM. Structural defects as a consequence of late intrauterine constraint, craniotabes, loose skin and asymmetric ear size. Semin Perinatol 1983;7:270.
- S. Butler MG et al: A 1S-item checklist for screening mentally retarded males for the fragile X syndrome. Clin Genet 1991;39:347-354.
- Hagerman RJ et al: Institutional screening for the fragile X syndrome. Am J Dis Child 1988;142:1216-1221.
- Gorlin RJ, Cohen MM, Hennekam RCM. Syndromes of the head and neck. 4th edition, 2001. Oxford University Press, New York.
- Feingold M, Bossert WH. Normal values for selected physical parameters: an aid to syndrome delineation. Birth Defects Orig Artic Ser 1974;10 (13):1–16.
Polyotia
Mirror imaging auricular duplications (1-3).
Polyotia is an extremely rare congenital anomaly of the external ear. There is mirror-image duplication of the auricle (Figure 23). The duplicated portion of the affected ear is positioned anteriorly and measures approximately half the size of the posterior, dysplastic ear. The entire complex has no visible external auditory meatus and no visible portions of the normal auricle or EAM.
According to Otto (1), these “cheek ears” (melotia) and so-called supernumerary pinna (polyotia) are to be regarded as unusually large ear tags. True pinna doubling is extremely rare. It is suggested that distinction between accessory appendages and true polyotia is debatable if one assumes that auricular appendages are duplicated structures of the auricular hillocks in the first place.
References
- Otto HD Pathogenesis of the preauricular appendages, melotia, and poliotia. Arch Otorhinolaryngol 1979;225(1):45-56.
- Bendor-Samuel Rl, Tung IC, Chen YR: Polyotia. Ann Plast Surg 1995;34:650.
- Ku PK, Tong MC, Yue V: Polyotia. Pediatr Otorhinolaryngol 1998;46:117.
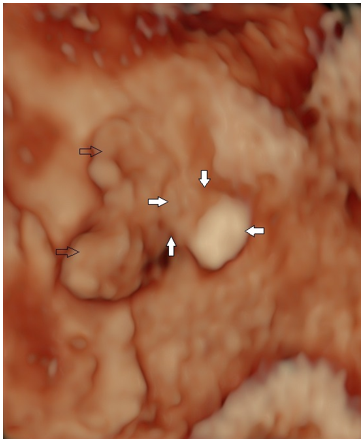
Figure 23. Polyotia
Dysplastic ear (open black arrows) with attempted duplication – periarticular tag adjacent to the ear (closed arrows).
References
- Otto HD. Pathogenese der branchiogenen Überschussbildungen. HNO Praxis 1983;8:161-9, 247-57.
This article should be cited as Suchet I, Santos J: Abnormalities of the auricle’s size and number. Visual Encyclopedia of Ultrasound in Obstetrics and Gynecology, www.isuog.org, June 2023.
Leave feedback or submit an image
We rely on your feedback to update and improve VISUOG. Please use the form below to submit any comments or feedback you have on this chapter.
If you have any images that you think would make a good addition to this chapter, please also submit them below - you will be fully credited for all images used.
Feedback form
Please note that the maximum upload size is 5MB, and larger images and video clips can be sent to [email protected].